Mapping between coordinate systems
Mapping coordinate systems: cheatsheet
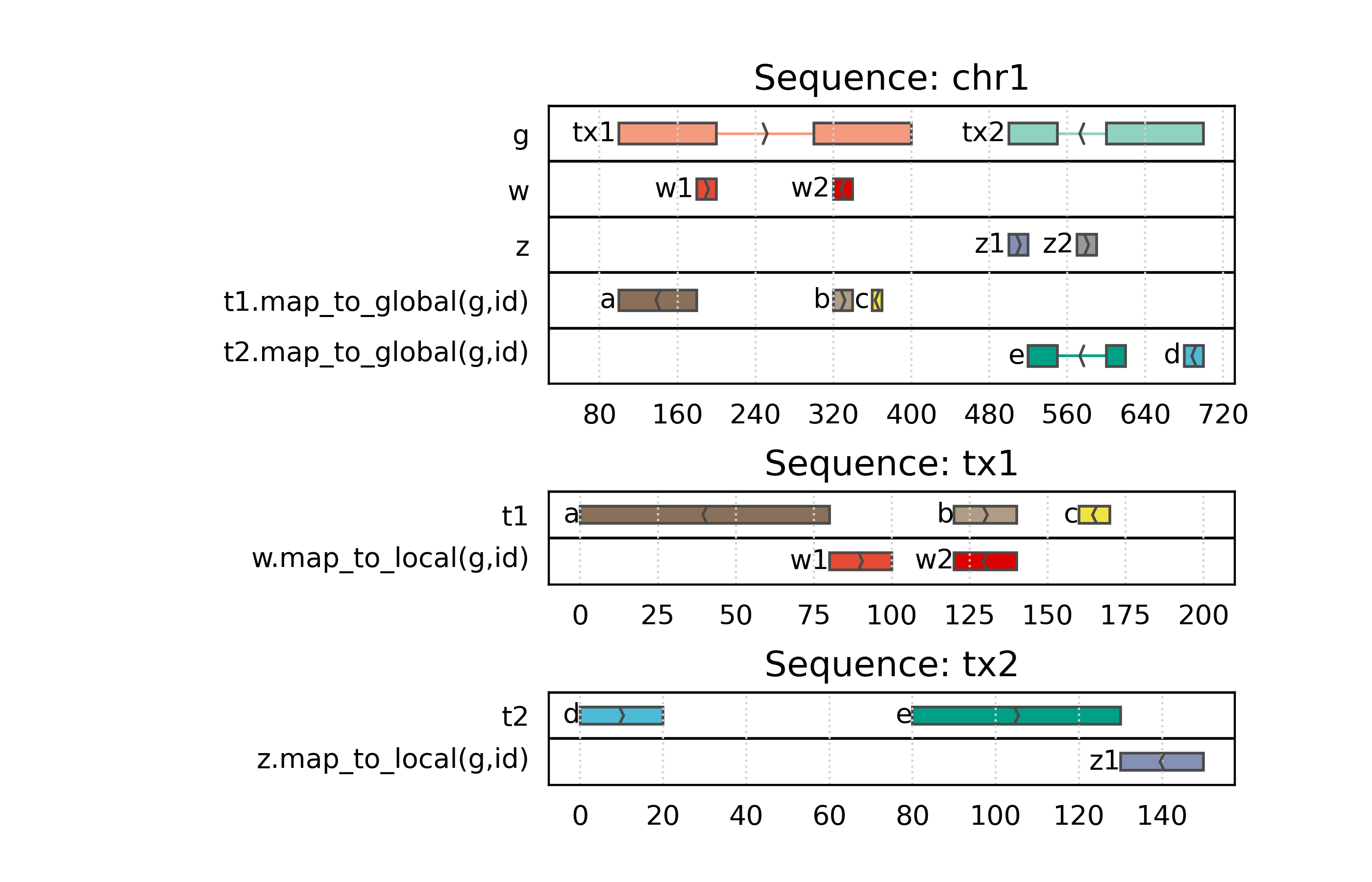
overlap.
Nested coordinate systems
PyRanges may represent nested coordinate systems, where the intervals in one coordinate system are relative to the intervals in another coordinate system. This is useful for representing hierarchical data.
Let’s make an intuitive case. On the one hand, we have some global ranges, that is genomic intervals wherein coordinates refer to positions along the genome:
>>> import pyranges1 as pr, pandas as pd
>>> gr = pr.example_data.ncbi_gff
>>> gre = (gr[gr.Feature=='exon']).get_with_loc_columns('Parent')
>>> gre
index | Chromosome Start End Strand Parent
int64 | category int64 int64 category str
------- --- ----------------- ------- ------- ---------- ---------------------
3 | CAJFCJ010000053.1 4882 5264 - rna-DGYR_LOCUS13733
7 | CAJFCJ010000053.1 10474 10958 + rna-DGYR_LOCUS13734
8 | CAJFCJ010000053.1 11028 11169 + rna-DGYR_LOCUS13734
9 | CAJFCJ010000053.1 11227 11400 + rna-DGYR_LOCUS13734
... | ... ... ... ... ...
141 | CAJFCJ010000025.1 2753 2851 - rna-DGYR_LOCUS12552-2
142 | CAJFCJ010000025.1 2593 2693 - rna-DGYR_LOCUS12552-2
143 | CAJFCJ010000025.1 2354 2537 - rna-DGYR_LOCUS12552-2
144 | CAJFCJ010000025.1 1909 2294 - rna-DGYR_LOCUS12552-2
PyRanges with 57 rows, 5 columns, and 1 index columns.
Contains 3 chromosomes and 2 strands.
On the other hand, we have local data, i.e. mapped to the transcript sequences that are annotated in the global ranges.
Below, we load some example data that we obtained by running the Rfam database of RNA motifs against the
transcript sequences in the gre object, using the software Infernal:
>>> rh = pr.example_data.rfam_hits
>>> rh = rh[ ['target_name', 'seq_from', 'seq_to', 'strand', 'query_name', 'mdl_from', 'mdl_to'] ]
>>> rh.head()
target_name seq_from seq_to strand query_name mdl_from mdl_to
0 rna-DGYR_LOCUS12552 267 311 + GAIT 1 71
1 rna-DGYR_LOCUS12552-2 288 332 + GAIT 1 71
2 rna-DGYR_LOCUS13738 1641 1678 + REN-SRE 1 37
3 rna-DGYR_LOCUS14091 137 43 - IFN_gamma 1 169
4 rna-DGYR_LOCUS14091 547 616 + snoZ30 1 97
Above, the target_name column contains the transcript IDs, which are
the same as the ID column in the gre object. Then we have coordinates that define the alignment between
portions of those transcripts and one of the RNA motifs (query models) in the Rfam database.
Let’s convert the rh object to a PyRanges object, taking care of switching from 1-based to 0-based coordinates:
>>> rh = rh.rename(columns={'target_name':'Chromosome', 'seq_from':'Start', 'seq_to':'End', 'strand':'Strand'})
>>> rh = pr.PyRanges(rh)
>>> rh['Start'] -= 1 # convert to 0-based coordinates
>>> rh
index | Chromosome Start End Strand query_name mdl_from mdl_to
int64 | str int64 int64 str str int64 int64
------- --- --------------------- ------- ------- -------- ------------ ---------- --------
0 | rna-DGYR_LOCUS12552 266 311 + GAIT 1 71
1 | rna-DGYR_LOCUS12552-2 287 332 + GAIT 1 71
2 | rna-DGYR_LOCUS13738 1640 1678 + REN-SRE 1 37
3 | rna-DGYR_LOCUS14091 136 43 - IFN_gamma 1 169
... | ... ... ... ... ... ... ...
31 | rna-DGYR_LOCUS13737 549 600 + mir-3047 1 61
32 | rna-DGYR_LOCUS13737 543 605 + mir-3156 1 77
33 | rna-DGYR_LOCUS13737 529 612 + MIR1523 1 92
34 | rna-DGYR_LOCUS13737 549 600 + MIR8001 1 67
PyRanges with 35 rows, 7 columns, and 1 index columns.
Contains 13 chromosomes and 2 strands.
Invalid ranges:
* 15 intervals are empty or negative length (end <= start). See indexes: 3, 5, 6, ...
Infernal, like other programs, reports negative stranded hits with Start and End reversed. Let’s fix that:
>>> rh.loc[ rh.Strand == '-', ['Start', 'End'] ] = rh.loc[ rh.Strand == '-', ['End', 'Start'] ].values
>>> rh
index | Chromosome Start End Strand query_name mdl_from mdl_to
int64 | str int64 int64 str str int64 int64
------- --- --------------------- ------- ------- -------- ------------ ---------- --------
0 | rna-DGYR_LOCUS12552 266 311 + GAIT 1 71
1 | rna-DGYR_LOCUS12552-2 287 332 + GAIT 1 71
2 | rna-DGYR_LOCUS13738 1640 1678 + REN-SRE 1 37
3 | rna-DGYR_LOCUS14091 43 136 - IFN_gamma 1 169
... | ... ... ... ... ... ... ...
31 | rna-DGYR_LOCUS13737 549 600 + mir-3047 1 61
32 | rna-DGYR_LOCUS13737 543 605 + mir-3156 1 77
33 | rna-DGYR_LOCUS13737 529 612 + MIR1523 1 92
34 | rna-DGYR_LOCUS13737 549 600 + MIR8001 1 67
PyRanges with 35 rows, 7 columns, and 1 index columns.
Contains 13 chromosomes and 2 strands.
Now we have the gre and rh objects, that represent global and local coordinate systems, respectively.
Let’s check that all Chromosome values in rh matches the ID column in gre:
>>> bool( rh.Chromosome.isin(gre.Parent).all() )
True
Mapping from local to global ranges
Next, we want to take the Rfam hits in rh, which are relative (local) to the transcript sequences,
and remap them to the genome (global) coordinates. To do so, we make use of the information in the gre object,
which defines the coordinates of each transcript, often split in exons, relative to the genome.
For this operation, we use the map_to_global method.
Besides the two PyRanges objects, we also need to specify the columns in the global range
that contains the identifier used as the Chromosome in the local range, provided by the global_on argument.
The resulting PyRanges object, rhg, contains the Rfam hits remapped to the genome coordinates:
>>> rhg = rh.map_to_global(gre, global_on='Parent')
>>> rhg
index | Chromosome Start End Strand query_name mdl_from mdl_to
int64 | category int64 int64 str str int64 int64
------- --- ----------------- ------- ------- -------- ------------ ---------- --------
0 | CAJFCJ010000025.1 2598 2643 - GAIT 1 71
1 | CAJFCJ010000025.1 2598 2643 - GAIT 1 71
2 | CAJFCJ010000053.1 77544 77582 + REN-SRE 1 37
3 | CAJFCJ010000097.1 2291 2384 - IFN_gamma 1 169
... | ... ... ... ... ... ... ...
31 | CAJFCJ010000053.1 39708 39759 + mir-3047 1 61
32 | CAJFCJ010000053.1 39702 39764 + mir-3156 1 77
33 | CAJFCJ010000053.1 39688 39771 + MIR1523 1 92
34 | CAJFCJ010000053.1 39708 39759 + MIR8001 1 67
PyRanges with 38 rows, 7 columns, and 1 index columns (with 3 index duplicates).
Contains 3 chromosomes and 2 strands.
Note that the transcript identifiers are now missing. To keep them, we can use the keep_id argument.
Analogously, we can record the local coordinates by using the keep_loc argument:
>>> rh.map_to_global(gre, global_on='Parent', keep_id=True, keep_loc=True).drop(
... columns=['query_name', 'mdl_from','mdl_to']) # dropping some columns to allow display
index | Chromosome Start End Strand Parent Start_local End_local ...
int64 | category int64 int64 str str int64 int64 ...
------- --- ----------------- ------- ------- -------- --------------------- ------------- ----------- -----
0 | CAJFCJ010000025.1 2598 2643 - rna-DGYR_LOCUS12552 266 311 ...
1 | CAJFCJ010000025.1 2598 2643 - rna-DGYR_LOCUS12552-2 287 332 ...
2 | CAJFCJ010000053.1 77544 77582 + rna-DGYR_LOCUS13738 1640 1678 ...
3 | CAJFCJ010000097.1 2291 2384 - rna-DGYR_LOCUS14091 43 136 ...
... | ... ... ... ... ... ... ... ...
31 | CAJFCJ010000053.1 39708 39759 + rna-DGYR_LOCUS13737 549 600 ...
32 | CAJFCJ010000053.1 39702 39764 + rna-DGYR_LOCUS13737 543 605 ...
33 | CAJFCJ010000053.1 39688 39771 + rna-DGYR_LOCUS13737 529 612 ...
34 | CAJFCJ010000053.1 39708 39759 + rna-DGYR_LOCUS13737 549 600 ...
PyRanges with 38 rows, 8 columns, and 1 index columns (with 3 index duplicates). (1 columns not shown: "Strand_local").
Contains 3 chromosomes and 2 strands.
Let’s now map intervals relative to protein sequences to genome coordinates. First, we obtain the coding sequences (CDS) from the original GFF file, and translate them to protein sequences (see Working with sequences for details):
>>> grc = (gr[gr.Feature=='CDS']).get_with_loc_columns('ID')
>>> genome_file = pr.example_data.files['ncbi.fasta']
>>> cds_seq = grc.get_sequence(genome_file, group_by='ID').str.upper()
>>> pep_seq = pr.seqs.translate(cds_seq)
>>> pep_seq.head()
ID
cds-CAD5125114.1 MSRQSGRSNDPRKVSGELLTLTYGALVAQLVKDSESDDEVNKQLDK...
cds-CAD5125115.1 MGYNIGIRLIEDFLARSSIGKCKDLRETAEIISKNGFKMFLNITPI...
cds-CAD5126491.1 MAKNPEKMSATKKLETINRCMGHTKRGLENGCYTKGLIKIRCFTAE...
cds-CAD5126492.1 MKIFAIISIYFILSESCYFRNVEVEGDFYLATFLAFHTDEYCTGPI...
cds-CAD5126493.1 MNFYRNFFNLIFCIKVSSFSPIQDYISCQEALTKTEQDGSYSIKPR...
Name: Sequence, dtype: object
As an example of a positional feature mapped to protein sequences, let’s find all instances of the amino acid ‘K’ (lysine):
>>> aa='K'
>>> z = [(seq_id, i, aa) for seq_id, seq in pep_seq.items() for i, char in enumerate(seq) if char == aa]
>>> z = pd.DataFrame(z, columns=['ID', 'Start', 'AminoAcid'])
>>> z.head()
ID Start AminoAcid
0 cds-CAD5125114.1 12 K
1 cds-CAD5125114.1 31 K
2 cds-CAD5125114.1 41 K
3 cds-CAD5125114.1 45 K
4 cds-CAD5125114.1 66 K
- Let’s convert to a PyRanges object:
>>> aa_pos = pr.PyRanges(z.rename(columns={'ID':'Chromosome'}).assign(End=lambda df: df.Start + 1 )) >>> aa_pos index | Chromosome Start AminoAcid End int64 | str int64 str int64 ------- --- ---------------- ------- ----------- ------- 0 | cds-CAD5125114.1 12 K 13 1 | cds-CAD5125114.1 31 K 32 2 | cds-CAD5125114.1 41 K 42 3 | cds-CAD5125114.1 45 K 46 ... | ... ... ... ... 457 | cds-CAD5126878.1 334 K 335 458 | cds-CAD5126878.1 341 K 342 459 | cds-CAD5126878.1 342 K 343 460 | cds-CAD5126878.1 350 K 351 PyRanges with 461 rows, 4 columns, and 1 index columns. Contains 17 chromosomes.
Next, we have to convert protein-based positions to nucleotide-based positions, still relative to the CDS. Because they’re in pythonic 0-based coordinates, we just need to multiply by 3:
>>> cds_pos = aa_pos.copy()
>>> cds_pos['Start'] *= 3
>>> cds_pos['End'] *= 3
Now we’re ready to map these positions to the genome coordinates. Let’s also fetch their underlying nucleotide sequence:
>>> genome_pos = cds_pos.map_to_global(grc, global_on='ID', keep_id=True)
>>> genome_pos['Sequence'] = genome_pos.get_sequence(genome_file).str.upper()
>>> genome_pos
index | Chromosome Start AminoAcid End ID Strand Sequence
int64 | category int64 str int64 str category object
------- --- ----------------- ------- ----------- ------- ---------------- ---------- ----------
0 | CAJFCJ010000025.1 3114 K 3117 cds-CAD5125114.1 - AAA
1 | CAJFCJ010000025.1 2797 K 2800 cds-CAD5125114.1 - AAG
2 | CAJFCJ010000025.1 2767 K 2770 cds-CAD5125114.1 - AAA
3 | CAJFCJ010000025.1 2755 K 2758 cds-CAD5125114.1 - AAA
... | ... ... ... ... ... ... ...
457 | CAJFCJ010000097.1 53008 K 53011 cds-CAD5126878.1 + AAG
458 | CAJFCJ010000097.1 53341 K 53344 cds-CAD5126878.1 + AAA
459 | CAJFCJ010000097.1 53344 K 53347 cds-CAD5126878.1 + AAA
460 | CAJFCJ010000097.1 53368 K 53371 cds-CAD5126878.1 + AAA
PyRanges with 466 rows, 7 columns, and 1 index columns (with 5 index duplicates).
Contains 3 chromosomes and 2 strands.
Because protein mapping to genome coordinates is common, map_to_map_to_global
provides a shortcut for this operation, through its pep_to_cds argument, which effectively multiplies the local
coordinates by 3 before mapping to the global coordinates. So this is equivalent to the previous operation:
>>> aa_pos.map_to_global(grc, global_on='ID', keep_id=True, pep_to_cds=True)
index | Chromosome Start AminoAcid End ID Strand
int64 | category int64 str int64 str category
------- --- ----------------- ------- ----------- ------- ---------------- ----------
0 | CAJFCJ010000025.1 3114 K 3117 cds-CAD5125114.1 -
1 | CAJFCJ010000025.1 2797 K 2800 cds-CAD5125114.1 -
2 | CAJFCJ010000025.1 2767 K 2770 cds-CAD5125114.1 -
3 | CAJFCJ010000025.1 2755 K 2758 cds-CAD5125114.1 -
... | ... ... ... ... ... ...
457 | CAJFCJ010000097.1 53008 K 53011 cds-CAD5126878.1 +
458 | CAJFCJ010000097.1 53341 K 53344 cds-CAD5126878.1 +
459 | CAJFCJ010000097.1 53344 K 53347 cds-CAD5126878.1 +
460 | CAJFCJ010000097.1 53368 K 53371 cds-CAD5126878.1 +
PyRanges with 466 rows, 6 columns, and 1 index columns (with 5 index duplicates).
Contains 3 chromosomes and 2 strands.
In the genetic code, the codons for lysine are ‘AAA’ or ‘AAG’, which fits what we see. One last important observation: note the warning above about index duplicates. Let’s take a look at them:
>>> genome_pos[genome_pos.index.duplicated(keep=False)]
index | Chromosome Start AminoAcid End ID Strand Sequence
int64 | category int64 str int64 str category object
------- --- ----------------- ------- ----------- ------- ---------------- ---------- ----------
234 | CAJFCJ010000053.1 77393 K 77395 cds-CAD5126496.1 + AA
234 | CAJFCJ010000053.1 77458 K 77459 cds-CAD5126496.1 + G
282 | CAJFCJ010000053.1 89719 K 89721 cds-CAD5126498.1 - AA
282 | CAJFCJ010000053.1 89660 K 89661 cds-CAD5126498.1 - G
... | ... ... ... ... ... ... ...
422 | CAJFCJ010000097.1 52381 K 52382 cds-CAD5126877.1 + A
422 | CAJFCJ010000097.1 52446 K 52448 cds-CAD5126877.1 + AG
446 | CAJFCJ010000097.1 52381 K 52382 cds-CAD5126878.1 + A
446 | CAJFCJ010000097.1 52446 K 52448 cds-CAD5126878.1 + AG
PyRanges with 10 rows, 7 columns, and 1 index columns (with 5 index duplicates).
Contains 2 chromosomes and 2 strands.
This is because the codon for some amino acids are split between two exons. In more general terms, this is an effect of mapping local features to a global coordinate system: if entities (e.g. CDS) encompasses multiple non-contiguous intervals in the global coordinate system (e.g. exons), a certain local feature (e.g. amino acid) may also be mapped into split intervals in global coordinates. These are identified by the index duplicates in the PyRanges object, as above.
Mapping from global to local ranges
Another task is to map from global to local ranges, which is the opposite of the previous task. This is useful when we put together data generated at different levels. For example, we may have genomic features predicted using the full genome, and we want to see where they reside in transcripts. In this example, we will map the sequence ‘AATAAA’, which is a polyadenylation signal motif, in genome sequences, then map them to transcript coordinates.
>>> pattern='AATAAA'
Let’s remind ourselves of the gre object, which contains the transcript coordinates:
>>> gre
index | Chromosome Start End Strand Parent
int64 | category int64 int64 category str
------- --- ----------------- ------- ------- ---------- ---------------------
3 | CAJFCJ010000053.1 4882 5264 - rna-DGYR_LOCUS13733
7 | CAJFCJ010000053.1 10474 10958 + rna-DGYR_LOCUS13734
8 | CAJFCJ010000053.1 11028 11169 + rna-DGYR_LOCUS13734
9 | CAJFCJ010000053.1 11227 11400 + rna-DGYR_LOCUS13734
... | ... ... ... ... ...
141 | CAJFCJ010000025.1 2753 2851 - rna-DGYR_LOCUS12552-2
142 | CAJFCJ010000025.1 2593 2693 - rna-DGYR_LOCUS12552-2
143 | CAJFCJ010000025.1 2354 2537 - rna-DGYR_LOCUS12552-2
144 | CAJFCJ010000025.1 1909 2294 - rna-DGYR_LOCUS12552-2
PyRanges with 57 rows, 5 columns, and 1 index columns.
Contains 3 chromosomes and 2 strands.
Let’s get the lengths of all these chromosomes from the genome:
>>> import pyfaidx
>>> pyf = pyfaidx.Fasta(genome_file)
>>> chromsizes = {chrom: len(pyf[chrom]) for chrom in gre.chromosomes}
>>> chromsizes
{'CAJFCJ010000025.1': 3418, 'CAJFCJ010000053.1': 109277, 'CAJFCJ010000097.1': 78757}
Let’s now create a PyRanges object than spans their whole sequence, with two intervals per chromosome: one for plus strand and one for minus strand; and let’s load their sequence in memory:
>>> q = [(chrom, 0, chromsizes[chrom], '+') for chrom in chromsizes] + [(chrom, 0, chromsizes[chrom], '-') for chrom in chromsizes]
>>> q
[('CAJFCJ010000025.1', 0, 3418, '+'), ('CAJFCJ010000053.1', 0, 109277, '+'), ('CAJFCJ010000097.1', 0, 78757, '+'), ('CAJFCJ010000025.1', 0, 3418, '-'), ('CAJFCJ010000053.1', 0, 109277, '-'), ('CAJFCJ010000097.1', 0, 78757, '-')]
>>> full_seq = pr.PyRanges( pd.DataFrame(q, columns=['Chromosome', 'Start', 'End', 'Strand']))
>>> full_seq['Sequence'] = full_seq.get_sequence(genome_file).str.upper()
>>> matches = []
>>> for i in full_seq.itertuples():
... st=0
... while (pos_match:= i.Sequence.find(pattern, st)) != -1:
... pat_st, pat_end = pos_match, pos_match+len(pattern)
... if i.Strand == '-':
... pat_st, pat_end = i.End - (pos_match + len(pattern)), i.End - pos_match
... matches.append( (i.Chromosome, pat_st, pat_end, i.Strand) )
... st=pos_match+1
>>> matches = pr.PyRanges( pd.DataFrame(matches, columns=['Chromosome', 'Start', 'End', 'Strand']))
>>> matches['Sequence'] = matches.get_sequence(genome_file).str.upper() # fetch seq as control
>>> matches
index | Chromosome Start End Strand Sequence
int64 | str int64 int64 str object
------- --- ----------------- ------- ------- -------- ----------
0 | CAJFCJ010000025.1 81 87 + AATAAA
1 | CAJFCJ010000025.1 90 96 + AATAAA
2 | CAJFCJ010000025.1 347 353 + AATAAA
3 | CAJFCJ010000025.1 553 559 + AATAAA
... | ... ... ... ... ...
619 | CAJFCJ010000097.1 2513 2519 - AATAAA
620 | CAJFCJ010000097.1 2284 2290 - AATAAA
621 | CAJFCJ010000097.1 2163 2169 - AATAAA
622 | CAJFCJ010000097.1 858 864 - AATAAA
PyRanges with 623 rows, 5 columns, and 1 index columns.
Contains 3 chromosomes and 2 strands.
The object matches contains the positions of the motif in the genome.
Now, we want to map these positions to the transcript coordinates. Function
map_to_local does exactly this.
For each motif, we need to decide which transcript we want to use as reference
coordinate system. By default, it will use all transcripts that overlap the motif:
>>> gre_matches = matches.map_to_local(gre, ref_on='Parent')
>>> gre_matches
index | Chromosome Start End Strand Sequence
int64 | str int64 int64 str object
------- --- ------------------- ------- ------- -------- ----------
28 | rna-DGYR_LOCUS13734 1142 1148 + AATAAA
29 | rna-DGYR_LOCUS13734 1416 1422 + AATAAA
30 | rna-DGYR_LOCUS13734 2106 2112 + AATAAA
31 | rna-DGYR_LOCUS13734 2232 2238 + AATAAA
... | ... ... ... ... ...
617 | rna-DGYR_LOCUS14091 980 986 - AATAAA
618 | rna-DGYR_LOCUS14091 774 780 - AATAAA
619 | rna-DGYR_LOCUS14091 265 271 - AATAAA
620 | rna-DGYR_LOCUS14091 36 42 - AATAAA
PyRanges with 59 rows, 5 columns, and 1 index columns (with 2 index duplicates).
Contains 14 chromosomes and 2 strands.
åAs before, you have options to retain metadata, in this case from the global ranges:
>>> matches.map_to_local(gre, ref_on='Parent', keep_chrom=True, keep_loc=True).columns
Index(['Chromosome', 'Start', 'End', 'Strand', 'Sequence', 'Chromosome_global',
'Start_global', 'End_global', 'Strand_global'],
dtype='str')
Let’s look at another use case. For example, we way have CDS and exon coordinates, and we want to map the CDS coordinates to the exon coordinates. In other words, we want to know where CDS intervals reside along the full mRNA transcripts.
Let’s inspect some data inside the gr object, focusing on a single identifier:
>>> pr.options.set_option('max_rows_to_show', 20)
>>> some_id = 'rna-DGYR_LOCUS12552-2'
>>> gr[gr.Parent == some_id].get_with_loc_columns(['Feature', 'Parent'])
index | Chromosome Start End Strand Feature Parent
int64 | category int64 int64 category category str
------- --- ----------------- ------- ------- ---------- ---------- ---------------------
140 | CAJFCJ010000025.1 3111 3250 - exon rna-DGYR_LOCUS12552-2
141 | CAJFCJ010000025.1 2753 2851 - exon rna-DGYR_LOCUS12552-2
142 | CAJFCJ010000025.1 2593 2693 - exon rna-DGYR_LOCUS12552-2
143 | CAJFCJ010000025.1 2354 2537 - exon rna-DGYR_LOCUS12552-2
144 | CAJFCJ010000025.1 1909 2294 - exon rna-DGYR_LOCUS12552-2
145 | CAJFCJ010000025.1 3111 3153 - CDS rna-DGYR_LOCUS12552-2
146 | CAJFCJ010000025.1 2753 2851 - CDS rna-DGYR_LOCUS12552-2
147 | CAJFCJ010000025.1 2593 2693 - CDS rna-DGYR_LOCUS12552-2
148 | CAJFCJ010000025.1 2354 2537 - CDS rna-DGYR_LOCUS12552-2
149 | CAJFCJ010000025.1 2174 2294 - CDS rna-DGYR_LOCUS12552-2
PyRanges with 10 rows, 6 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.
You can see that CDS and exon belonging to the same transcript have the same Parent value.
Therefore, we can easily map the CDS coordinates to the exon coordinates of the corresponding transcript,
by pairing (overlapping) CDS and exon intervals with the same Parent value:
>>> pr.options.reset_options()
>>> cds = gr[gr.Feature == 'CDS'].get_with_loc_columns(['Feature', 'Parent'])
>>> exons = gr[gr.Feature == 'exon'].get_with_loc_columns(['Feature', 'Parent'])
>>> cds_local = cds.map_to_local(exons, ref_on='Parent', match_by='Parent')
>>> cds_local
index | Chromosome Start End Strand Feature
int64 | str int64 int64 str category
------- --- --------------------- ------- ------- -------- ----------
0 | rna-DGYR_LOCUS13733 1 382 + CDS
1 | rna-DGYR_LOCUS13734 258 484 + CDS
2 | rna-DGYR_LOCUS13734 484 625 + CDS
3 | rna-DGYR_LOCUS13734 625 798 + CDS
... | ... ... ... ... ...
52 | rna-DGYR_LOCUS12552-2 139 237 + CDS
53 | rna-DGYR_LOCUS12552-2 237 337 + CDS
54 | rna-DGYR_LOCUS12552-2 337 520 + CDS
55 | rna-DGYR_LOCUS12552-2 520 640 + CDS
PyRanges with 56 rows, 5 columns, and 1 index columns.
Contains 17 chromosomes and 1 strands.
>>> cds_local[cds_local.Chromosome == some_id]
index | Chromosome Start End Strand Feature
int64 | str int64 int64 str category
------- --- --------------------- ------- ------- -------- ----------
51 | rna-DGYR_LOCUS12552-2 97 139 + CDS
52 | rna-DGYR_LOCUS12552-2 139 237 + CDS
53 | rna-DGYR_LOCUS12552-2 237 337 + CDS
54 | rna-DGYR_LOCUS12552-2 337 520 + CDS
55 | rna-DGYR_LOCUS12552-2 520 640 + CDS
PyRanges with 5 rows, 5 columns, and 1 index columns.
Contains 1 chromosomes and 1 strands.
With the similar logic, we can easily map the start and stop codon positions to the exon coordinates:
>>> cds.slice_ranges(0, 3).assign(Feature='start').map_to_local(exons, ref_on='Parent', match_by='Parent')
index | Chromosome Start End Strand Feature
int64 | str int64 int64 str str
------- --- --------------------- ------- ------- -------- ---------
0 | rna-DGYR_LOCUS13733 1 4 + start
1 | rna-DGYR_LOCUS13734 258 261 + start
2 | rna-DGYR_LOCUS13734 484 487 + start
3 | rna-DGYR_LOCUS13734 625 628 + start
... | ... ... ... ... ...
52 | rna-DGYR_LOCUS12552-2 139 142 + start
53 | rna-DGYR_LOCUS12552-2 237 240 + start
54 | rna-DGYR_LOCUS12552-2 337 340 + start
55 | rna-DGYR_LOCUS12552-2 520 523 + start
PyRanges with 56 rows, 5 columns, and 1 index columns.
Contains 17 chromosomes and 1 strands.
>>> cds.slice_ranges(-3).assign(Feature='stop').map_to_local(exons, ref_on='Parent', match_by='Parent')
index | Chromosome Start End Strand Feature
int64 | str int64 int64 str str
------- --- --------------------- ------- ------- -------- ---------
0 | rna-DGYR_LOCUS13733 379 382 + stop
1 | rna-DGYR_LOCUS13734 481 484 + stop
2 | rna-DGYR_LOCUS13734 622 625 + stop
3 | rna-DGYR_LOCUS13734 795 798 + stop
... | ... ... ... ... ...
52 | rna-DGYR_LOCUS12552-2 234 237 + stop
53 | rna-DGYR_LOCUS12552-2 334 337 + stop
54 | rna-DGYR_LOCUS12552-2 517 520 + stop
55 | rna-DGYR_LOCUS12552-2 637 640 + stop
PyRanges with 56 rows, 5 columns, and 1 index columns.
Contains 17 chromosomes and 1 strands.